Joseph Gish1, Shaun Peters1, Kenneth Childers1, Ian Smith1, Ernest Parker2, John Healey2, H. Trent Spencer2, Chris Doering2, Pete Lollar2 and P. Clint Spiegel, Jr.1
1Department of Chemistry – Western Washington University, Bellingham, WA, USA; 2Aflac Cancer and Blood Disorders Center, Department of Pediatrics – Emory University, Atlanta, GA, USA
Abstract
The most common epitopes for neutralizing anti-factor VIII (fVIII) antibodies (inhibitors) are localized to the A2 and C2 domains, which are shared amongst both acquired hemophilia A and congenital hemophilia A patients. These inhibitors commonly interfere with binding of fVIII to activated factor IXa, platelet membrane surfaces (PS) and von Willebrand factor (vWF). By contrast, recent evidence indicates that the C1 domain contributes to the inhibitor response, potentially disrupting the ability of fVIII to bind PS and/or vWF, blocking the endocytosis of fVIII by antigen presenting cells, or serving as a weak inhibitor that rapidly accelerates fVIII clearance. Epitope localization for anti-fVIII inhibitors has largely been characterized through ELISA-based detection with human/porcine chimeric fVIII hybrids or H/D exchange protection patterns with LC-MS/MS detection. Previous structural findings of antibody epitopes have been limited to complexes between anti-C2 domain inhibitors and the isolated fVIII C2 domain. In this study, we have determined the structure of an anti-C1 domain pathogenic inhibitor (2A9) in complex with a bioengineered construct of fVIII (ET3i). The 2A9 inhibitor shows weak inhibition of fVIII binding to both PS and vWF, and mutational analysis localizes the epitope to F2068, which overlaps with anti-C1 antibodies present in hemophilia A patients. This structure shows that the epitope directly interacts with F2068 in the C1 domain, but also makes specific contacts with the adjacent A3 domain. Overall, this study represents the first antibody complex with an intact construct of B domain-deleted fVIII and lends structural evidence to the anti-C1 domain pathogenic immune response.
Procoagulant Activity of Factor VIII
Factor VIII (fVIII) is a procoagulant glycoprotein that resides in circulation bound tightly to von Willebrand Factor (VWF)1. Upon initiation of the blood coagulation cascade, fVIII is activated by thrombin or fXa (to form fVIIIa), which dissociates from VWF and subsequently binds to fIXa in the presence of the negatively charged phospholipids that are presented on the surface of activated platelets (Figure 1)2. The fVIIIa/fIXa complex is known as the intrinsic ‘tenase’ complex, which also converts fX to fXa. The activated form of fVIII (fVIIIa) functions as a localization and conformational cofactor for the active fIXa serine protease, increasing the Vmax of the tenase reaction by ~200,000-fold2. Without sufficient procoagulant activity from fVIII, hemostasis cannot be maintained.

Figure 1: The Blood Coagulation Cascade. Two pathways (extrinsic and intrinsic) that are initiated by the exposure of tissue factor (TF) or PS groups of activated platelet membranes to circulating protein factors, respectively. (diamonds: inactive proenzymes, circles: active serine proteases)
Factor VIII Structure
Factor VIII is an essential, non-enzymatic cofactor that performs a central role in the regulation of blood coagulation3. The full-length, unprocessed fVIII protein is synthesized as a monomer of 2,332 amino acid residues and has the domain structure: A1-A2-B-A3-C1-C24-7 (Figure 2). The inactive form of FVIII resides in circulation bound tightly to VWF (KD ~ 0.5 nM) as a heterodimer of a heavy chain (A1-A2-B) and light chain (A3-C1-C2)1,8. Once proteolytically activated, fVIIIa dissociates from VWF as a heterotrimeric assembly (A1/A2/A3-C1-C2).

Figure 2: Domain structure of factor VIII. Factor VIII circulates as a heterodimer bound to VWF. Upon activation, factor VIII dissociates as a trimeric assembly. Red – inhibitory antibodies target different regions of factor VIII
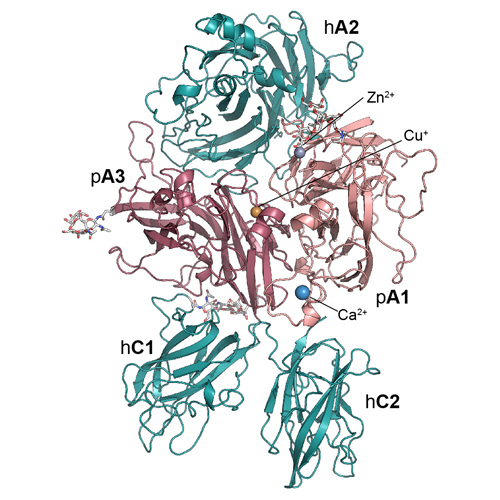
In recent work, we report the refined molecular structure of the ET3i human/porcine chimeric FVIII construct at 3.2 Å, the highest resolution structure of FVIII communicated to date (Figure 3). Briefly, a chimeric construct, designated “ET3i” (previously denoted as HP47), containing the porcine A1 and A3 domains, demonstrates higher expression and activity levels than recombinant hFVIII in in vitro and in vivo expression systems such as those used for recombinant FVIII manufacturing and clinical gene therapy, respectively.9-18 This human/porcine chimera provides insight to the inter-domain surface contact residues that may contribute to the differential stability of human and porcine FVIII proteins. Furthermore, a novel conformational domain movement is observed between the C2 domains of the two ET3i molecules in the crystallographic asymmetric unit (ASU). Based on the role of the C2 domain in binding to phosphatidylserine-rich activated platelet surfaces, this observed conformational movement could have physiological implications to the membrane binding of FVIII. Together with complementary biochemical studies, the C1 and C2 domains are thought to be primarily responsible for membrane association19,20.

Figure 3: The structure of the bioengineered ET3i. (A) Ribbon diagram representation of the ET3i A model (PDBID#: 6MF0, pink: porcine A1 domain, dark magenta: porcine A3 domain, dark teal: human A2, C1 and C2 domains). (B) Domain structure of ET3i (shaded region: porcine sequence, unshaded region: human sequence). (C) Asymmetric unit of ET3i. Two molecules are present within the asymmetric unit and modeled independently (dark teal: model A, purple: model B). (D) superposition of ET3i A model with the previous X-ray crystal structure of human FVIII (PDBID#: 2R7E, dark teal: ET3i A model, yellow: human FVIII).
The C2 domain of fVIII was previously determined as essential for the interaction of fVIII with VWF (via the VWF D’D3 domain), and fVIIIa with activated platelet surfaces (via the phosphatidylserine polar headgroup)21. These interactions are mutually exclusive, indicating the binding sites overlap22-24. We have developed a refined model for membrane association by the fVIII C2 domain, which we previously proposed based on high-resolution X-ray structures of antibody inhibitor complexes with the isolated C2 domain25 (Figure 4). Here, a set of solvent-exposed hydrophobic residues protrudes from b-hairpin turns that extend from the core structure, which favorably insert into the nonpolar interior of the plasma membrane. An additional ring of basic residues provides a positively charged surface directly adjacent to the hydrophobic residues for favorable charge-charge interactions with the negatively charged polar head group of phosphatidylserine, which centers on R2320. This model of membrane association is not completely consistent with other structural and biochemical data26-29, and it must be refined further. Moreover, new data indicate that the C1 domain contributes to membrane binding and is a critical domain for VWF binding and fVIII cofactor function19,20,30-32.

Figure 4. Factor VIII C2 domain-specific inhibitory antibody epitopes and membrane binding models. (A) Ribbon diagram representation of the fVIII C2 domain bound to different classes of C2-specific inhibitory antibodies. Each X-ray crystal structure was superimposed with the C2 domain structure and residues involved in the different epitopes are shown by stick representation (red: fVIII C2 domain; green: G99 mAb, a non-classical BC epitope; blue/cyan: 3E6 mAb, a classical A epitope; yellow: BO2C11 mAb, a classical AB epitope). (B) Proposed PS membrane binding models for the fVIII C2 domain (left: old PS binding model, including the non-classical epitope with residues K2227 and V2223; right: new PS binding model, including both 3E6 and BO2C11 classical antibody epitopes, which centers at residue R2320).
Development of Antibody Inhibitors
Therapy for hemophilia A is most commonly infusion of either plasma-derived or recombinant fVIII33-35. Today, the most significant complication of fVIII replacement therapy is the development of fVIII inhibitory antibodies, appearing in approximately 30% of patients with hemophilia A that receive treatment36-39. Further, rare spontaneous development of fVIII inhibitor antibodies in non-hemophilia patients also occurs, resulting in acquired hemophilia40. The epitopes recognized by inhibitor antibodies for fVIII that specifically block its function (both in vitro and in vivo) most commonly reside within the A2 and C2 domains41, and new data indicate that antibodies against the C1 domain are also pathogenic42-44. Historically, fVIII antibody inhibitors have been thought to block fVIII procoagulant function by a variety of mechanisms: (1) Block the ability of fVIII to bind and activate factors IXa and X; (2) Inhibit fVIII binding to VWF and/or activated platelet surfaces45-47; or (3) hinder the activation of fVIII by thrombin (or fXa) and/or subsequent release of fVIII from VWF48-50. “Classical” anti-C2 antibodies function by inhibiting the binding of fVIIIa to the negatively charged surfaces that resemble activated platelets. Our recent structural work has elucidated the epitopes for both “classical” and “non-classical” antibody inhibitors against the C2 domain, and these studies have informed the refining of our membrane binding model for the C2 domain, which we extrapolate to how fVIII binds platelet surfaces28,51 (Figure 4).
The Immune Response to the C1 Domain
Recent evidence has shown that inhibitors develop specifically against the C1 domain. With a murine hemophilia A model, inhibitory anti-C1 domain antibodies have been characterized that harbor epitopes that block the ability of fVIII to bind activated platelet surfaces, von Willebrand factor, and block endocytosis by antigen presenting cells, which represents the three main functions of the C1 domain in circulation44. Moreover, several of these antibodies show direct competition with the antibody responses present in both acquired and congenital hemophilia A patients. One monoclonal antibody under study, 2A9, is a group A type inhibitor that modestly blocks the ability of fVIII to bind both activated platelet surfaces and von Willebrand factor (Table 1)44. This particular antibody has been shown to have an overlapping epitope with 60% of patient inhibitor samples, is commonly present in acquired hemophilia A patients, and is pathogenic in a mouse model. Mutational analysis has indicated that the 2A9 epitope contains residue F2068, which is part of a loop that is also recognized by hemophilia A patients with inhibitors52.

Table 2: X-ray Data collection and refinement statistics. Statistics for the highest-resolution shell are shown in parentheses.


Figure 5: Structure of ET3i with anti-C1 2A9 inhibitor antibody. (A) Crystal structure of ET3i:2A9 complex. fVIII domains and 2A9 heavy chain (HC) and light chain (LC) are colored. (B) Interfacial contacts between A3 and C1 domains and 2A9 are a mixture of hydrophobic and hydrophilic interactions. Mutations to F2068 and R2150 are reported to abolish 2A9 binding.

Figure 6: Comparison of reported binding sites for 2A9 and the D’D3 domains of vWF on the C1 domain. (A) C1 domain (orange) from our ET3i:2A9 structure. (B and C) Hydrogen-deuterium exchange mass spectrometry results overlaid on C1 domain structures from Batsuli et al. and Chiu et al., respectively. Arrows depict reported binding sites for 2A9 or D’D3.

Figure 7. Disruption of ET3i:D’D3 complex by 2A9. (Top) Anti-His tips were loaded with His6-D’D3 and measured ET3i association and dissociation (red). We then prepared the ET3i:2A9 complex and found no measurable binding (green). Background was measured from anti-His6 tips lacking D’D3 (blue). (Bottom) Anti-mouse IgG tips were loaded with 2A9 and measured ET3i association and dissociation (magenta). We then prepared the ET3i:D’D3 complex and ~50% loss in affinity (gold). Background was measured from anti-mouse IgG tips lacking 2A9 (green).

Figure 8. Sedimentation assay with isolated C1 domain and ET3i with 2A9. The isolated C1 domain (top) or ET3i (bottom) were incubated with lipid membranes in the absence or presence of 2A9. Samples were collected from the pellet (P) and supernatant (SN) and analyzed using SDS-PAGE. The C1 domain, heavy chain (ET3iHC, 2A9HC) and light chain (ET3iLC, 2A9LC) are indicated by arrows.

Figure 9. Conformational changes of the ET3i C1 and C2 domains when bound to 2A9. Structural alignment of our ET3i:2A9 to unbound ET3i reveals perturbations to the C1 and C2 domains. While the C1 domain shows only modest conformational changes when bound to 2A9, the C2 domain adopts a novel conformation that includes a ~17 Å shift to the platelet membrane-binding loops. The 2A9 epitope on the C1 domain is highlighted in green.

NIH U54 HL141981
NIH R15 HL135658
References
- Foster PA, Fulcher CA, Marti T, Titani K, Zimmerman TS. A major factor VIII binding domain resides within the amino-terminal 272 amino acid residues of von Willebrand factor. J Biol Chem. 1987;262(18):8443-8446.
- van Dieijen G, Tans G, Rosing J, Hemker HC. The role of phospholipid and factor VIIIa in the activation of bovine factor X. J Biol Chem. 1981;256(7):3433-3442.
- Lawson JH, Kalafatis M, Stram S, Mann KG. A model for the tissue factor pathway to thrombin. I. An empirical study. J Biol Chem. 1994;269(37):23357-23366.
- Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature. 1984;312(5992):326-330.
- Kane WH, Davie EW. Blood coagulation factors V and VIII: structural and functional similarities and their relationship to hemorrhagic and thrombotic disorders. Blood. 1988;71(3):539-555.
- Christophe OD, Lenting PJ, Kolkman JA, Brownlee GG, Mertens K. Blood coagulation factor IX residues Glu78 and Arg94 provide a link between both epidermal growth factor-like domains that is crucial in the interaction with factor VIII light chain. J Biol Chem. 1998;273(1):222-227.
- Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature. 1984;312(5992):342-347.
- Hill-Eubanks DC, Parker CG, Lollar P. Differential proteolytic activation of factor VIII-von Willebrand factor complex by thrombin. Proc Natl Acad Sci U S A. 1989;86(17):6508-6512.
- Dooriss KL, Denning G, Gangadharan B, et al. Comparison of factor VIII transgenes bioengineered for improved expression in gene therapy of hemophilia A. Hum Gene Ther. 2009;20(5):465-478.
- Spencer HT, Denning G, Gautney RE, et al. Lentiviral vector platform for production of bioengineered recombinant coagulation factor VIII. Mol Ther. 2011;19(2):302-309.
- Brown HC, Wright JF, Zhou S, et al. Bioengineered coagulation factor VIII enables long-term correction of murine hemophilia A following liver-directed adeno-associated viral vector delivery. Mol Ther Methods Clin Dev. 2014;1:14036.
- Brown HC, Zakas PM, George SN, Parker ET, Spencer HT, Doering CB. Target-Cell-Directed Bioengineering Approaches for Gene Therapy of Hemophilia A. Mol Ther Methods Clin Dev. 2018;9:57-69.
- Doering CB, Denning G, Dooriss K, et al. Directed engineering of a high-expression chimeric transgene as a strategy for gene therapy of hemophilia A. Mol Ther. 2009;17(7):1145-1154.
- Doering CB, Denning G, Shields JE, et al. Preclinical Development of a Hematopoietic Stem and Progenitor Cell Bioengineered Factor VIII Lentiviral Vector Gene Therapy for Hemophilia A. Hum Gene Ther. 2018;29(10):1183-1201.
- Doering CB, Healey JF, Parker ET, Barrow RT, Lollar P. Identification of porcine coagulation factor VIII domains responsible for high level expression via enhanced secretion. J Biol Chem. 2004;279(8):6546-6552.
- Spencer HT, Riley BE, Doering CB. State of the art: gene therapy of haemophilia. Haemophilia. 2016;22 Suppl 5:66-71.
- Tran R, Myers DR, Denning G, et al. Microfluidic Transduction Harnesses Mass Transport Principles to Enhance Gene Transfer Efficiency. Mol Ther. 2017;25(10):2372-2382.
- Zakas PM, Brown HC, Knight K, et al. Enhancing the pharmaceutical properties of protein drugs by ancestral sequence reconstruction. Nat Biotechnol. 2017;35(1):35-37.
- Hsu TC, Pratt KP, Thompson AR. The factor VIII C1 domain contributes to platelet binding. Blood. 2008;111(1):200-208.
- Lu J, Pipe SW, Miao H, Jacquemin M, Gilbert GE. A membrane-interactive surface on the factor VIII C1 domain cooperates with the C2 domain for cofactor function. Blood. 2011;117(11):3181-3189.
- Spiegel PC, Jr., Stoddard BL. Optimization of factor VIII replacement therapy: can structural studies help in evading antibody inhibitors? Br J Haematol. 2002;119(2):310-322.
- Andersson LO, Brown JE. Interaction of factor VIII-von Willebrand Factor with phospholipid vesicles. Biochem J. 1981;200(1):161-167.
- Lajmanovich A, Hudry-Clergeon G, Freyssinet JM, Marguerie G. Human factor VIII procoagulant activity and phospholipid interaction. Biochim Biophys Acta. 1981;678(1):132-136.
- Healey JF, Lubin IM, Nakai H, et al. Residues 484-508 contain a major determinant of the inhibitory epitope in the A2 domain of human factor VIII. J Biol Chem. 1995;270(24):14505-14509.
- Pratt KP, Shen BW, Takeshima K, Davie EW, Fujikawa K, Stoddard BL. Structure of the C2 domain of human factor VIII at 1.5 A resolution. Nature. 1999;402(6760):439-442.
- Brison CM, Mullen SM, Wuerth ME, et al. The 1.7 A X-ray crystal structure of the porcine factor VIII C2 domain and binding analysis to anti-human C2 domain antibodies and phospholipid surfaces. PLoS One. 2015;10(3):e0122447.
- Meeks SL, Healey JF, Parker ET, Barrow RT, Lollar P. Antihuman factor VIII C2 domain antibodies in hemophilia A mice recognize a functionally complex continuous spectrum of epitopes dominated by inhibitors of factor VIII activation. Blood. 2007;110(13):4234-4242.
- Walter JD, Werther RA, Brison CM, et al. Structure of the factor VIII C2 domain in a ternary complex with 2 inhibitor antibodies reveals classical and nonclassical epitopes. Blood. 2013;122(26):4270-4278.
- Walter JD, Werther RA, Polozova MS, et al. Characterization and solution structure of the factor VIII C2 domain in a ternary complex with classical and non-classical inhibitor antibodies. J Biol Chem. 2013;288(14):9905-9914.
- Bloem E, van den Biggelaar M, Wroblewska A, et al. Factor VIII C1 domain spikes 2092-2093 and 2158-2159 comprise regions that modulate cofactor function and cellular uptake. J Biol Chem. 2013;288(41):29670-29679.
- Wakabayashi H, Fay PJ. Replacing the factor VIII C1 domain with a second C2 domain reduces factor VIII stability and affinity for factor IXa. J Biol Chem. 2013;288(43):31289-31297.
- Wakabayashi H, Griffiths AE, Fay PJ. Factor VIII lacking the C2 domain retains cofactor activity in vitro. J Biol Chem. 2010;285(33):25176-25184.
- Hoots WK. The future of plasma-derived clotting factor concentrates. Haemophilia. 2001;7 Suppl 1:4-9.
- Mauser-Bunschoten EP, van der Bom JG, Bongers M, et al. Purity of factor VIII product and incidence of inhibitors in previously untreated patients with haemophilia A. Haemophilia. 2001;7(4):364-368.
- Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in haemophilia A patients–a review of recent studies of recombinant and plasma-derived factor VIII concentrates. Haemophilia. 1999;5(3):145-154.
- Bray GL, Gomperts ED, Courter S, et al. A multicenter study of recombinant factor VIII (recombinate): safety, efficacy, and inhibitor risk in previously untreated patients with hemophilia A. The Recombinate Study Group. Blood. 1994;83(9):2428-2435.
- Kreuz W, Ettingshausen CE, Zyschka A, et al. Inhibitor development in previously untreated patients with hemophilia A: a prospective long-term follow-up comparing plasma-derived and recombinant products. Semin Thromb Hemost. 2002;28(3):285-290.
- Lusher JM, Arkin S, Abildgaard CF, Schwartz RS. Recombinant factor VIII for the treatment of previously untreated patients with hemophilia A. Safety, efficacy, and development of inhibitors. Kogenate Previously Untreated Patient Study Group. N Engl J Med. 1993;328(7):453-459.
- Lusher JM, Lee CA, Kessler CM, Bedrosian CL, ReFacto Phase 3 Study G. The safety and efficacy of B-domain deleted recombinant factor VIII concentrate in patients with severe haemophilia A. Haemophilia. 2003;9(1):38-49.
- Kessler CM, Knobl P. Acquired haemophilia: an overview for clinical practice. Eur J Haematol. 2015;95 Suppl 81:36-44.
- Prescott R, Nakai H, Saenko EL, et al. The inhibitor antibody response is more complex in hemophilia A patients than in most nonhemophiliacs with factor VIII autoantibodies. Recombinate and Kogenate Study Groups. Blood. 1997;89(10):3663-3671.
- Healey JF, Parker ET, Barrow RT, Langley TJ, Church WR, Lollar P. The comparative immunogenicity of human and porcine factor VIII in haemophilia A mice. Thromb Haemost. 2009;102(1):35-41.
- Wroblewska A, van Haren SD, Herczenik E, et al. Modification of an exposed loop in the C1 domain reduces immune responses to factor VIII in hemophilia A mice. Blood. 2012;119(22):5294-5300.
- Batsuli G, Deng W, Healey JF, et al. High-affinity, noninhibitory pathogenic C1 domain antibodies are present in patients with hemophilia A and inhibitors. Blood. 2016;128(16):2055-2067.
- Jacquemin MG, Desqueper BG, Benhida A, et al. Mechanism and kinetics of factor VIII inactivation: study with an IgG4 monoclonal antibody derived from a hemophilia A patient with inhibitor. Blood. 1998;92(2):496-506.
- Shima M, Nakai H, Scandella D, et al. Common inhibitory effects of human anti-C2 domain inhibitor alloantibodies on factor VIII binding to von Willebrand factor. Br J Haematol. 1995;91(3):714-721.
- Shima M, Scandella D, Yoshioka A, et al. A factor VIII neutralizing monoclonal antibody and a human inhibitor alloantibody recognizing epitopes in the C2 domain inhibit factor VIII binding to von Willebrand factor and to phosphatidylserine. Thromb Haemost. 1993;69(3):240-246.
- Nogami K, Shima M, Hosokawa K, et al. Factor VIII C2 domain contains the thrombin-binding site responsible for thrombin-catalyzed cleavage at Arg1689. J Biol Chem. 2000;275(33):25774-25780.
- Nogami K, Shima M, Hosokawa K, et al. Role of factor VIII C2 domain in factor VIII binding to factor Xa. J Biol Chem. 1999;274(43):31000-31007.
- Saenko EL, Shima M, Gilbert GE, Scandella D. Slowed release of thrombin-cleaved factor VIII from von Willebrand factor by a monoclonal and a human antibody is a novel mechanism for factor VIII inhibition. J Biol Chem. 1996;271(44):27424-27431.
- Spiegel PC, Jr., Jacquemin M, Saint-Remy JM, Stoddard BL, Pratt KP. Structure of a factor VIII C2 domain-immunoglobulin G4kappa Fab complex: identification of an inhibitory antibody epitope on the surface of factor VIII. Blood. 2001;98(1):13-19.
- Kahle J, Orlowski A, Stichel D, et al. Frequency and epitope specificity of anti-factor VIII C1 domain antibodies in acquired and congenital hemophilia A. Blood. 2017;130(6):808-816.