
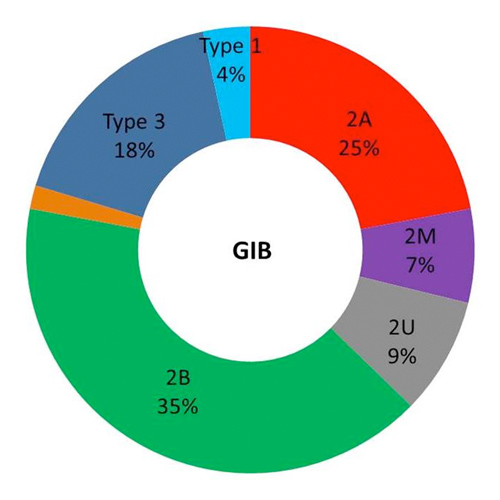
Exploration and treatment strategies for gastrointestinal bleeding in Von Willebrand Disease over a 3-year period: a french-nationwide retrospective survey
METHODS A 15-items mail-based survey was first sent to 30 centers between December 2017 and February 2018 to identify constitutional VWD-patients referred for at least one GIB episode (including hematemesis, melena, hematochezia or unexplained chronic iron deficiency...
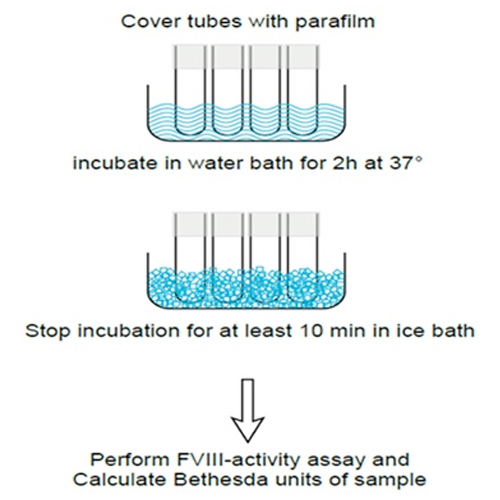
Use of chromogenic Factor VIII activity determination in Haemophilia A plasma of patients under emicizumab treatment
Introduction Factor VIII activity (FVIII:C) assays of samples containing recombinant FVIII or the new bispecific antibody emicizumab (Haemlibra®) can be associated with differences in FVIII recovery in vitro between one-stage-clotting-assays and chromogenic assays....

Performance evaluation of a new fully automated thrombin generation instrument for the measurement of TGA in Haemophilia samples
Introduction Background To monitor treatment in haemophilic patients is crucial. It is well known that global assays or FVIII/FIX activity determination do not correlate with bleeding episodes. For patients treated with B domain deleted FVIII preparation (rFVIII) the...
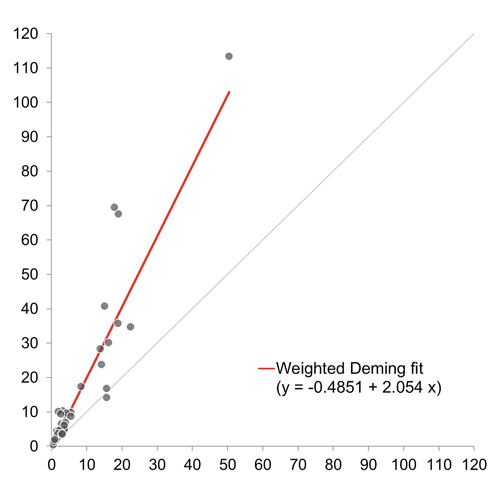
ADAMTS13 Inhibitor Assessment with the HemosIL AcuStar ADAMTS13 Activity Assay
BACKGROUND Thrombotic thrombocytopenic purpura (TTP) is a rare hematological disease characterized by the severe deficiency of ADAMTS13 activity. In acquired TTP, reduced ADAMTS13 activity is caused by autoantibodies inhibiting ADAMTS13 function (i.e., ADAMTS13...
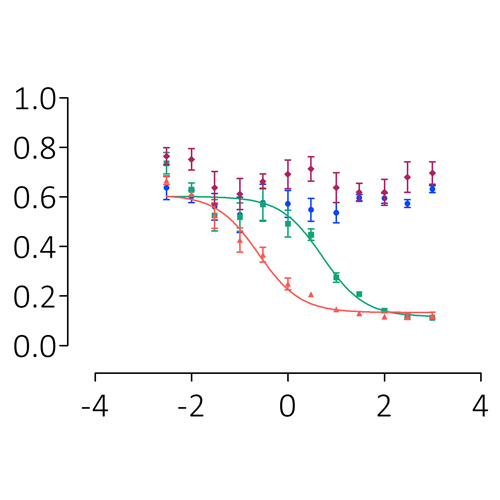
Recombinant, patient-derived FVIII-neutralising antibodies: a platform for research, product testing, and ex vivo modelling of haemophilia A
Background Congenital haemophilia A (HA) is a rare, sex-linked, hereditary condition that results in mild, moderate, or severe bleeding due to a deficiency of endogenous factor VIII (FVIII). Exogenous FVIII can be administered to restore coagulation but 30% of severe...

Non-specific non-antiphospholipid inhibitor in a 26-years-old woman: a case report with few answers and many questions
A 26-years old woman was admitted to the ambulatory of internal medicine the "Miulli" Hospital of the city of Acquaviva delle Fonti (Bari) for hypochromic microcytic anemia, with hemoglobin levels of 7 g/L and severe iron deficiency. The patient was in good health...
Long-term neuropsychological sequelae, emotional wellbeing and quality of life in patients with acquired thrombotic thrombocytopenic purpura
BACKGROUND Acquired thrombotic thrombocytopenic purpura (TTP) is a rare multisystem microangiopathy with fluctuating signs and symptoms, typically of the nervous system, caused by the immune-mediated severe deficiency of the von Willebrand factor-cleaving protease...
In vitro and in vivo modulation of von Willebrand factor gene mutations with dominant-negative effect
Backgroud Von Willebrand Disease (VWD) is the most common inherited bleeding disorder with a prevalence of 0.6 - 1.3% based on population studies. The severity of the disease is mostly determined by the level of residual von Willebrand factor (VWF) activity and the...
Thrombin and plasmin generation in patients with plasminogen or plasminogen activator inhibitor type 1 deficiency
Introduction: Deficiencies of plasminogen and plasminogen activator inhibitor type 1 (PAI-1) are rare disorders of fibrinolysis. Conventional laboratory assays of fibrinolysis lack specificity and validation. Moreover, there is a weak correlation of the assays with...
Quantification of VWF propeptide release before and after desmopressin in von Willebrand disease and hemophilia A
Background Von Willebrand factor (VWF) levels represent a balance between synthesis, secretion and clearance of VWF* VWF propeptide (VWFpp) is a marker of VWF synthesis Increased VWFpp/VWF:Ag ratio reflects increased VWF clearance Adding VWFpp to population...