Matteo Campioni1, Paulette Legendre2, Cécile Loubière2, Barbara Lunghi1, Mirko Pinotti1, Olivier D. Christophe2, Peter J. Lenting2, Cécile V. Denis2, Francesco Bernardi1, Caterina Casari2
1Department of Life Sciences and Biotechnology, University of Ferrara, Ferrara, Italy; 2Inserm UMR_S1176, Le Kremlin-Bicêtre, France; Univ Paris-Sud, Le Kremlin-Bicêtre, France
Backgroud
Von Willebrand Disease (VWD) is the most common inherited bleeding disorder with a prevalence of 0.6 – 1.3% based on population studies.
The severity of the disease is mostly determined by the level of residual von Willebrand factor (VWF) activity and the disease subtype. Partial or complete quantitative deficiencies are classified as VWD-type 1 and 3, respectively. VWD-type 2 groups all qualitative defects and it is divided in 4 subtypes: A, B, M and N. VWD is inherited as autosomal dominant traits in most of the cases except for VWD-type 3 and VWD-type 2N.
VWF is a large multimeric protein, which undergoes several finely regulated biosynthetic steps. Relevant for this project are the dimerization and the multimerization processes.
Due to the multimeric nature of the protein, heterozygous mutations occurring in the VWF gene, are susceptible to display dominant-negative effect. When a mutant VWF monomer associates with normal subunits the activity of the normal subunit is also affected, resulting in a more severe phenotype compared to a classic heterozygous mutation.
For this project two dominant-negative mutations are of interest. The first one is large in frame deletion that impairs multimerization while the second one is a mutation affecting the dimerization process.
The P1127_C1948delinsR deletion (DEL) results in a smaller monomer lacking all the A domains, part of the D3- and part of the D4-domain and has a new in-frame junction between exons 25 and 35 (an Arg, now at position 1127, replaces Cys 1948)

The mutation impairing dimerization is the missense Cys to Arg substitution at position 2773 in the cysteine knot (CK)-like domain.

Aim
The aim of our study was to better understand the interaction of dimerization and mutimerization mutations, and to design strategies to rescue the dominant-negative effect of the p.P1127_C1948delinsR deletion.
Methods
Two approaches were used.
The first approach consisted in combining both the missense mutation and the large deletion in a single VWF subunit.
In the normal situation, multimers are made of subunits originating from the two alleles. In both mutants, the multimerization process is interrupted upon the incorporation of the mutant subunit. The idea is that by completely preventing incorporation of the double mutant subunit, multimerization will be rescued and multimers of only wild-type subunits will be produced.

This approach was evaluated in vitro in COS cells and in vivo in Vwf-deficient mice receiving hydrodynamic gene transfer.
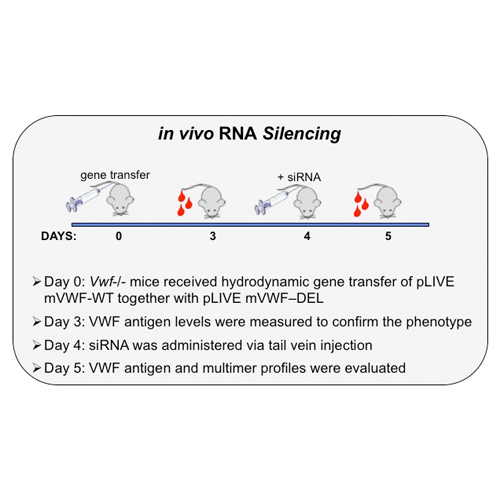
For the second approach, we used siRNA to selectively block expression of the mutant “allele” by targeting the junction sequence connecting exons 25 to 35, while the expression of the wild-type “allele” was unaffected.

This approach has been previously validated in vitro (Casari C. et al. Blood 2010) and was evaluated in vivo in Vwf-deficient mice receiving hydrodynamic gene transfer and siRNA treatment.
Approach #1

in vitro results

The combination of the missense and DEL mutations increased VWF antigen levels in plasma (left panel), although not significantly, and highly ameliorated the multimer profile. At low resolution (middle panel) a considerable increase of high molecular weight multimers was observed and at high resolution (right panel), we observed the complete disappearance of the DEL-band.
in vivo results

In vivo, the combination of the missense and the DEL mutation increased plasmatic VWF antigen levels (left panel) to about 50% of WT, which is expected when only one allele is expressed. This resulted in a big improvement of the multimer profile (middle panel) with restoration of normal high molecular weight multimers. Finally, when the bleeding phenotype was assessed in a tail-transection assay (right panel), we observed that mice expressing the DEL mutation had a severe phenotype, while mice expressing the two mutations displayed a bleeding tendency similar to that observed in mice expressing only WT-VWF.
Approach #2

in vivo results

The in vivo silencing approach resulted in a small but significant increase of VWF antigen levels (left panel) in plasma of mice receiving the siDEL (red), compared to non treated mice (grey). Multimer profiles were improved in two ways. First, some high molecular weight multimers were visible in the plasma of mice receiving siDEL (middle panel); and, second, the intensity of the band corresponding to the DEL-subunit was reduced in dimers, tetramers, hexamers and octamers of mice one day post siDEL treatment, compared to the intensity quantified before treatment.
Conclusions
Due to their ability to invalidate wild-type subunits, heterozygous dominant-negative mutations are generally associated with protein expression and activity below 50%, resulting in severe phenotypes. They represent a clinical challenge as they are difficult to diagnose and to treat.
In this project we demonstrated that molecular strategies can be applied to modulate dominant-negative mutations in order to correct their negative effect. These original approaches can be applied in vitro and in vivo to characterize and confirm the dominant effect of new mutations. Furthermore, we applied for the first time a siRNA-based approach to a mouse model of severe VWD.